PharmDr. Petr Horák
Nemocniční lékárna Fakultní nemocnice v Motole
Katedra lékárenství IPVZ
Úvod
Tzv. Orphan drugs jsou léčiva, která jsou určena pro léčbu vzácných onemocnění, jak ostatně vyjadřuje i označení této kategorie v české legislativě[1] - “léčivé přípravky pro vzácná onemocnění”.
Vývoj léčiv pro vzácná onemocnění je zásadní otázkou na jedné straně komerční, na straně druhé etickou. Snahou regulačních úřadů, které navrhují legislativu spojenou s orphan drugs, je zajistit takové podmínky pro výzkumné a výrobní společnosti, aby se jim výzkum v této oblasti vyplatil a bylo možné pro pacienty se vzácnými onemocněními léčiva získat.
Vzácná onemocnění
Definice vzácných onemocnění
Definice vzácných onemocnění je čistě arbitrární a jako taková se v jednotlivých částech světa liší. Většina zemí s rozvinutým systémem zdravotnictví již přijala legislativu pro orphan drugs a tato legislativa v sobě definici zahrnuje. Příklad pro EU[2], USA[3] a Japonsko[4] uvádí tabulka 1.
Tabulka 1.: Definice vzácných onemocnění v různých zemích
|
|
| Země | Definice vzácného onemocnění | | EU | Smrtící či chronicky invalidizující nemoc, kterou trpí méně než 5 z 10 000 obyvatel EU | | USA | Nemoc, kterou trpí méně než 200 000 obyvatel USA | | Japonsko | Nemoc, kterou trpí méně než 50 000 obyvatel státu |
|
|
|
Vzácná onemocnění z komplexního pohledu
Relativně nízká hranice prevalence pro zařazení nemoci mezi vzácná onemocnění neznamená, že je také malý počet pacientů, kteří vzácnými onemocněními trpí. Odhaduje se, že nyní je rozpoznáno asi 7 000 – 8 000 vzácných onemocnění. V přepočtu na populaci USA to značí, že jen ve Spojených Státech trpí vzácnými onemocněními asi 20 až 25 milionů obyvatel, v součtu s EU asi 55 milionů lidí. Celkem tedy asi 6 - 8 % obyvatel EU trpí některým vzácným onemocněním. Navíc je každý rok identifikováno přibližně 250 nových vzácných onemocnění.[5] Na tomto trendu se podílí mj. výsledky genetického a farmakogenetického výzkumu, který umožňuje odhalit základní příčiny jednotlivých vzácných onemocnění.
Většina vzácných onemocnění je vrozená, existují ale i vzácná onemocnění získaná. Nejčastěji se manifestují od dětského věku. Mezi nejčastěji se vyskytující vzácná onemocnění patří cystická fibróza, hemofilie všech typů, Duchenneova svalová dystrofie, Charcot-Marie-Toothova choroba, retinitis pigmentosa.
Orphan drugs
Historie orphan drugs
Kodifikaci orphan drugs do své legislativy provedly nejprve Spojené Státy, a to v roce 1983. Následovaly další země (viz tabulka 2), až konečně v roce 2000 byla přijata i legislativa evropská, konkrétně Nařízení rady 141/2000.[6]
|
|
| Tabulka 2: přijetí legislativy o orphan drugs v různých zemích
|
|
| Země | Rok přijetí legislativy | | USA | 1983 | | Japonsko | 1985 | | Singapur | 1997 | | Austrálie | 1998 | | EU | 2000 |
|
|
|
Od začátků lékové legislativy k Orphan drug Act
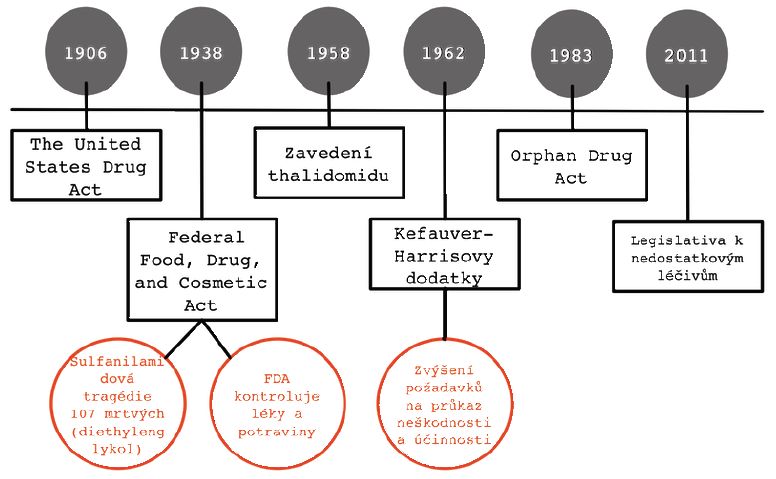
Od prvního zákona, který byl na federální úrovni k lékové problematice přijat (The United States Drug Act) do přijetí legislativy k orphan drugs uplynulo necelých 80 let. Tato doba byla charakteristická postupným zvyšováním nároků na bezpečnost a kvalitu léčiv a zkoušky provedené před jejich uvedením do praxe. První zásadní zpřísnění přišlo v roce 1938 s “Federal Food, Drug, and Cosmetic Act”, který byl reakcí na sulfanilamidovou tragédii a který mj. zavedl dohled FDA nad vývojem a zacházením s léčivy.Po přijetí Kefauver-Harrisových dodatků, jež byly reakcí na evropskou thalidomidovou tragédii, došlo k zásadnímu zpřísnění požadavků na předregistrační studie. To však významně navýšilo náklady na vstup léčiv do praxe a pro výrobce přestalo být atraktivní a ekonomicky únosné přicházet s novými léky pro
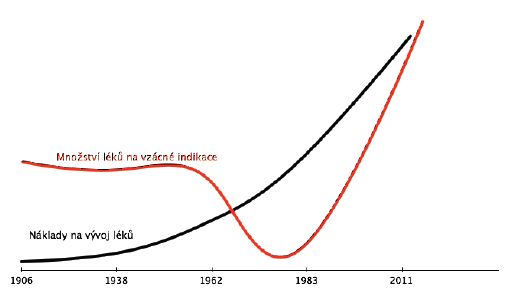
nemoci s nízkou prevalencí. Počet nových léků pro tyto indikace konstantně klesal a reakcí na tento fakt bylo právě přijetí příslušného zákona, tj. “The United States Orphan Drug Act” (viz obr. 1).[4] To vedlo k obrácení trendu a počet nových léků pro vzácné indikace začal zásadně stoupat, jak ilustruje graf 1.
|
|
|
Ilustrační graf: množství nových léků na vzácné indikace v čase (USA)
|
|
|
Obrázek 1: vývoj legislativy v oblasti léků v USA
pozn.: kliknutím se obrázek zvětší
Zajímavost - “Herceptin Case”
Přijetí Orphan Drug Act v USA vedlo k rychlému a strmému nárůstu počtu přihlášek k získání statutu orphan drug. Důvodem jsou poměrně velkorysé výhody, které přiznání tohoto statutu v USA nabízí:
- Daňové úlevy až do výše 50 % nákladů na klinický vývoj,
- přístup ke grantům,
- pomoc s administrací žádosti o registraci a zrychlení lhůt,
- odpuštění poplatků za podání žádosti o registraci, a především
- sedmiletá exkluzivita na trhu.
Je ještě na místě zdůraznit, že exkluzivita na trhu je nezávislá na patentové ochraně, a na rozdíl od ní běží až od okamžiku uvedení přípravku na trh. [7][8]
Tyto lákavé obchodní výhody vedou k velkému zájmu o získání statutu orphan drug mezi výrobci. Spolu s nastavením hranice pro orphan drug to vede k získání tohoto statutu i u léčiv, které např. v EU šanci na získání nemají.
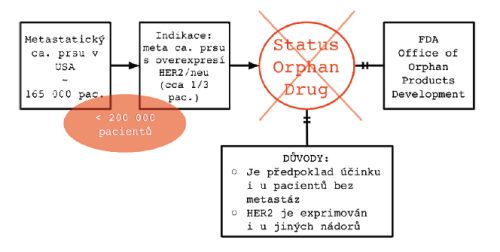
Příkladem může být tzv. “Herceptin case”, tedy způsob, jakým FDA zamítla žádost o přiznání statutu orphan drug pro Herceptin®.
Toto posuzování proběhlo v roce 1998 a ukázalo na řadů aspektů, které je třeba při posuzování orphan drugs brát do úvahy.
Žádost byla podána pro indikaci metastatického karcinomu prsu s overexpresí HER2/neu. Vzhledem k tomu, že prevalence metastatického karcinomu prsu v USA byla v té době cca 165 000 pacientů (a tedy méně než je hranice pro orphan drug) a pacientů s expresí HER2 je dokonce jen 1/3, zdálo by se přiznání statutu OD jasné.
FDA ale přiznání odmítla, a to proto, že neuznala definici cílové skupiny pacientů pouze pro metastatického karcinomu prsu meta s overexpresí HER2/neu. Zdůvodnila to následovně:
- Je předpoklad účinku i u pacientů bez metastáz.
- HER2 je exprimován i u jiných nádorů, a také u nich existuje pravděpodobnost účinku.
Tento případ ukazuje na jednu ze zásadních otázek v rámci problematiky orphan drugs: kde začíná a končí definice jednotlivého onemocnění a diagnózy? S nástupem stále přesnějších diagnostických metod umožňujících velmi přesné odlišení různých podtypů nemocí se samozřejmě snižuje i počet pacientů, kteří danými typy nemoci trpí.
|
|
|
Obrázek 2: “The Herceptin Case”
Na druhou stranu je třeba říci, že přes tento přístup FDA existuje v USA mnoho případů léků, které patří mezi orphan drugs a zároveň se staly tzv. block-bustery, tedy léčivy, které dosáhly ročního obratu vyššího než 1 mld. dolarů.
V současnosti je v USA na trhu necelých 400 léčiv ze skupiny orphan drugs, tedy podstatně více, než je orphan drugs v EU.[5][7]
Nové molekuly a “repositioning”
Statut orphan drug se neobjevuje pouze u nově vyvinutých léčiv. Byl udělen i některým existujícím léčivům, které byly schváleny původně pro jinou indikaci (např. použití amphotericinu u leishmaniózy, sildenafilu u plicní hypertenze), nebo dokonce u léčiv, které byly z trhu staženy (jako je tomu u thalidomidu pro použití u mnohočetného myelomu a lepry, či arsen trioxidu pro akutní promylocytární leukémii).
Toto “znovupoužití” často vyplyne ze spolupráce velkých firem s dalšími výzkumnými organizacemi, které screenují vlastnosti již zavedených
léčiv u vzácných indikací. Výrobce k tomu vede především snaha o optimální využití svého patentového portfolia.[9]
Vývoj nových molekul s indikacemi vzácných onemocnění samozřejmě nezůstává pozadu.
V USA je podána žádost o statut orphan drug až u čtvrtiny nových molekul.[7]
Orphan drugs v EU
Výhody pro výrobce orphan drugs v EU a dalších zemích
Všechny vyspělé země ve svých příslušných zákonech poskytují výhody společnostem , které orphan drugs vyvíjejí. Srovnání přináší tabulka 3 (dle [8]).
Tabulka 3: podpora vývoje orphan drugs v různých zemích
|
|
| Země | Výhody | | | | | | Exkluzivita na trhu | Daňové úlevy | Žádosti, poplatky | Další | | EU | 10 let | - | Osvobození od poplatků,
asistence s žádostí | Národní i evropské
podpůrné programy | | USA | 7 let | Do 50 % nákladu na klin. hodnocení | Osvobození od poplatků,
asistence s žádostí | Grantové programy | | Japonsko | 10 let | 6 % z veškerého výzkumu, do výše 10 % celkových daní firmy | - | Až 50 % finanční podpora
vývoje | | Austrálie | 5 let | Osvobození od poplatků | - | - |
|
|
|
Indikace orphan drugs v EU
Vzhledem k podstatně kratší historii a menšímu počtu žádostí o statut orphan drug je počet schválených orphan drugs v EU v souladu s nařízením Rady 141/2000 o dost menší, než je tomu v USA (k listopadu 2011 schváleno 62 léčiv, pro více než 75 diagnóz, a naopak více než 40 žádostí bylo neúspěšných).[10],[11] Přesto se samozřejmě jedná o značný nárůst oproti předchozímu stavu, kdy de facto na trhu existovalo pouhých 8 léčiv pro vzácná onemocnění.[12]
Orphan drugs mají nejčastěji indikace onkologické, kardiologické, neurologická a vrozená metabolická onemocnění.[10]
Náklady na orphan drugs
Vzhledem k velkému počtu pacientů trpících některým ze vzácných onemocnění a postupnému nárůstu počtu orphan drugs náklady na jejich použití v minulých letech rostly po přijetí nařízení 141/2000 o cca 10 % ročně. Predikce dalšího vývoje je poněkud složitá, a to díky značné variabilitě nákladů na jednoho nemocného u různých vzácných chorob.
Nyní se celkový odhad podílu na celkových výdajích za farmaceutické přípravky v EU odhaduje na 3,3 % s postupným očekávaným nárůstem na 4,6 % do roku 2016.[13]
Limity a výhody skupiny orphan drugs, závěr
Orphan drugs jako jako přístup k léčbě vzácných onemocněních jsou rozhodně nadějná. Jako problematické se jeví správné nastavení legislativy, aby statut orphan drugs získala jen ta léčiva, u nichž je to skutečně potřeba[5]. Existuje také problém postupně rostoucích nákladů, ačkoliv ten je zčásti vyvážen např. snížením nákladů na hospitalizaci atd.
Poměrně významným problémem také zůstává v řadě případů jen velmi limitovaná klinická zkušenost s většinou těchto přípravků v okamžiku, kdy se dostávají do praxe. Ta je pochopitelně způsobena jen omezenými možnostmi získání dostatečného počtu subjektů do klinických studií.[14]
|
|
Vysvětlivky
Cystická fibróza
Je vrozenou poruchou obvyklou především v kavkazské populaci, s odhadovanou prevalencí 1:8 000 až 1:10 000. Jde o recesivní autozomální onemocnění (chromozóm 7), které na molekulární úrovni spočívá v poruše CFTR proteinu (Cystic Fibrosis Transmembrane Conductance Regulator), který řídí pohyby hlavně chloridových iontů na membránách.
Onemocnění se v první řadě projevuje velmi slaným potem (s rizikem hyponatremické dehydratace) a produkcí velmi hustého hlenu v dýchacích cestách. Postižena může být řada vnitřních orgánů, hlavní manifestace je nicméně na dýchacích cestách a pankreatu. Jde o chronické a progredující onemocnění, díky značným pokrokům v léčbě je dnes očekávaná délka života pacientů 40 let.[15]
Hemofilie
Hemofilie je genetické onemocnění charakterizované spontánním krvácením způsobeným deficitem srážecího faktoru VIII (hemofilie A) nebo IX (hemofilie B). Má různou intenzitu podle míry deficitu srážecího faktoru, u vážné formy (pod 1 % normální aktivity) dochází k častým spontánním krvácením.
Onemocnění se nejčastěji projeví u dětí ve věku, kdy se učí chodit. Je onemocněním vázaným na X chromozóm, mírnější formy onemocnění se ale mohou někdy projevovat i žen – přenašeček.[16]
Vedle hemofilie A a B existuje i hemofilie C, která je způsobena vrozeným deficitem faktoru XI. Jde o onemocnění postihující obě pohlaví.[17]
Duchenneova svalová dystrofie
Duchenneova svalová dystrofie je vrozeným neuromuskulárním onemocněním charakterizovaná progresivní svalovou slabostí příčně pruhovaného, hladkého svalstva i myokardu. Je onemocněním mužů (incidence 1/3 300 porodů).[18]
|
|
Charcot-Marie-Toothova choroba
Charcot-Marie-Toothova choroba je nejčastější vrozenou neuropatií. Jedná se o celý komplex onemocnění, s řadou forem. Projevuje se postižením periferních nervů dolních i horních končetin s následným ochabováním svalstva a senzorického nervstva. Postihuje jednoho z 2 500 lidí.[19]
Retinitis pigmentosa
Retinitis pigmentosa je skupina vrozených chorob postihující jednoho z 3 000 - 7 000 lidí. Je charakterizována abnormalitami fotoreceptorů nebo epitelu sítnice a provázena progresivní ztrátou zraku. Může mít autozomální dominantní formu, autozomální recesivní formu nebo být vázána na chromozom X. Dosud bylo odhaleno přes 40 genů, které mají s onemocněním spojitost.[20]
|
|
Literatura:
1. Zákon č. 378/2007 Sb. o léčivech
2. Nařízení Rady (ES) č. 141/2000
3. The United States Orphan Drug Act
4. Taruscio, D., Capozzoli, F., & Frank, C. (2011). Rare diseases and orphan drugs. Ann Ist Super Sanita, 47(1), 83–93.
doi:10.4415/ANN_11_01_17
5. Wellman-Labadie, O., & Zhou, Y. (2010). The US Orphan Drug Act: rare disease research stimulator or commercial opportunity? Health Policy, 95(2–3), 216–228. doi:10.1016/j.healthpol.2009.12.001
6. Rogers, A. (2000). Europe finally agrees to encourage orphan-drug production. The Lancet, 8.
7. Freeman, S. N., Burke, K. A., Imoisili, M. A., & Cote, T. R. (2010). The Orphan Drug Act and the development of stem cell-based products for rare diseases. Cell Stem Cell, 7(3), 283–287. doi:10.1016/j.stem.2010.08.003
8. Visschers, R. G. J., van, G., Wim G., & Olde, D., Steven W.M. (2011). Orphan diseases neglected: The faith of intestinal failure. e-SPEN, the European e-Journal of Clinical Nutrition and Metabolism, 6(5), e232-e234. doi:10.1016/j.eclnm.2011.07.003
9. Muthyala, R. (2011). Orphan/rare drug discovery through drug repositioning. Drug Discovery Today: Therapeutic Strategies,.
doi:10.1016/j.ddstr.2011.10.003
10. Přehled schválených léčiv [online]. Dostupné na http://www.ema.europa.eu. Cit. 10. 12. 2011 (http://www.ema.europa.eu. Cit. 10. 12. 2011)
11. Putzeist, M., Heemstra, H. E., Garcia, J. L., Mantel-Teeuwisse, A. K., Wied, C. C. G.-D., Hoes, A. W., & Leufkens, H. G. M. (2011). Case study: determinants for successful marketing authorisation of orphan medicinal products in the EU. Drug Discovery Today,.
doi:10.1016/j.drudis.2011.10.027
12. Taylor, L.: EU orphan drug regulation “is a success” [online]. Pharma Times Online
(http://www.pharmatimes.com/Article/10-11-30/EU_orphan_drug_regulation_%e2%80%9cis_a_success%e2%80%9d.aspx), 30. 11. 2011 (cit. 11. 12. 2011).
13. Schey, C., Milanova, T., & Hutchings, A. (2011). Estimating the budget impact of orphan medicines in Europe: 2010 - 2020. Orphanet J Rare Dis, 6, 62. doi:10.1186/1750–1172–6–62
14. Dupont, A. G., & Van Wilder, P. B. (2011). Access to orphan drugs despite poor quality of clinical evidence. Br J Clin Pharmacol, 71(4),
488–496. doi:10.1111/j.1365–2125.2010.03877.x
15. Cystic fibrosis [Online]. Dostupné na Orphanet - klikněte zde
(cit. 4. 12. 2011)
16. Goudeman, J.: Hemophilia [online]. Dostupné na Orphanet - klikněte zde
(cit 4. 12. 2011)
17. Goudeman, J.: Congenital factor XI deficiency [online]. Dostupné na Orphanet - klikněte zde (cit. 4. 12.2011)
18. Quinlivan, R.: Duchenne muscular dystrophy [online]. Dostupné na: Orphanet - klikněte zde (cit. 4. 12. 2011) (cit. 4. 12. 2011)
19. Miller, L. J., Saporta, A. S., Sottile, S. L., Siskind, C. E., Feely, S. M., & Shy, M. E. (2011). Strategy for genetic testing in Charcot-Mariedisease. Acta Myol, 30(2), 109–116. Retrieved from - klikněte zde.
20. Ferrari, S., Di Iorio, E., Barbaro, V., Ponzin, D., Sorrentino, F. S., & Parmeggiani, F. (2011). Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics, 12(4), 238–249. doi:10.2174/138920211795860107
|
|
|